公众号/
编辑 | 萝卜皮
来自深度语言模型的蛋白质表征,已经在计算蛋白质工程的许多任务中表现出最先进的性能。近年来,进展主要集中在参数计数上,最近模型的容量超过了它们所训练的数据集的大小。
牛津大学(University of Oxford)的研究人员提出一个替代方向。他们证明,在密码子而不是氨基酸序列上训练的大型语言模型可以提供高质量的表征,并且在各种任务中都优于同类最先进的模型。
在某些任务中,例如物种识别、蛋白质和转录本丰度预测等,该团队发现,基于密码子训练的语言模型优于所有其他已发布的蛋白质语言模型,包括一些包含超过 50 倍训练参数的 模型 。
该研究以「Codon language embeddings provide strong signals for use in protein engineering」为题于 2024 年 2 月 23 日发布在《Nature Machine Intelligence》。

蛋白质表征学习仍存在不少挑战
预训练语言模型已成为计算蛋白质工程许多领域不可或缺的工具。大多数标记蛋白质数据集的大小有限,因此首先在大型、未标记的序列信息语料库(例如 UniRef)上对庞大的深度神经网络进行预训练,并具有自监督的重建目标。自监督训练赋予模型的潜在变量具有高度信息性的特征,称为表征学习,然后可以在可用训练数据有限的下游任务中利用这些特征。
蛋白质表征学习目前是用于预测变异适应性、蛋白质功能、亚细胞定位、溶解度、结合位点、信号肽、翻译后修饰、内在紊乱等的最先进工具的核心,它们在实现准确的免比对蛋白质结构预测的道路上显示出了巨大潜力。因此,改进学习表征是在计算蛋白质工程中实现一致、实质性改进的潜在途径。
迄今为止,实现更多信息表征的途径遵循两个主要方向:追求增强规模的模型,其中增加模型容量单调地提高性能;模型架构的改进也持续带来了性能提升。但是,这两个方向都耗费人力和计算机时间,需要显著优化,并且似乎提供递减(对数)回报。
更丰富的数据是另一条途径
改进学习表征的另一种途径可能是使用包含更丰富信号的生物数据。虽然蛋白质语言模型迄今为止主要关注氨基酸序列,但编码蛋白质的 DNA 序列中还包含其他信息。
蛋白质编码 DNA (cDNA) 的语言依赖于 64 个核苷酸三联体,称为密码子,每个密码子编码一个特定的氨基酸或序列的末端。
虽然这种 64 密码子字母表是高度简并的,大多数氨基酸由多达 6 个不同的密码子编码,但目前的研究表明,编码相同氨基酸(同义)的密码子不能互换使用。同义密码子的使用与蛋白质结构特征相关,近 60 个同义突变与人类疾病有关。
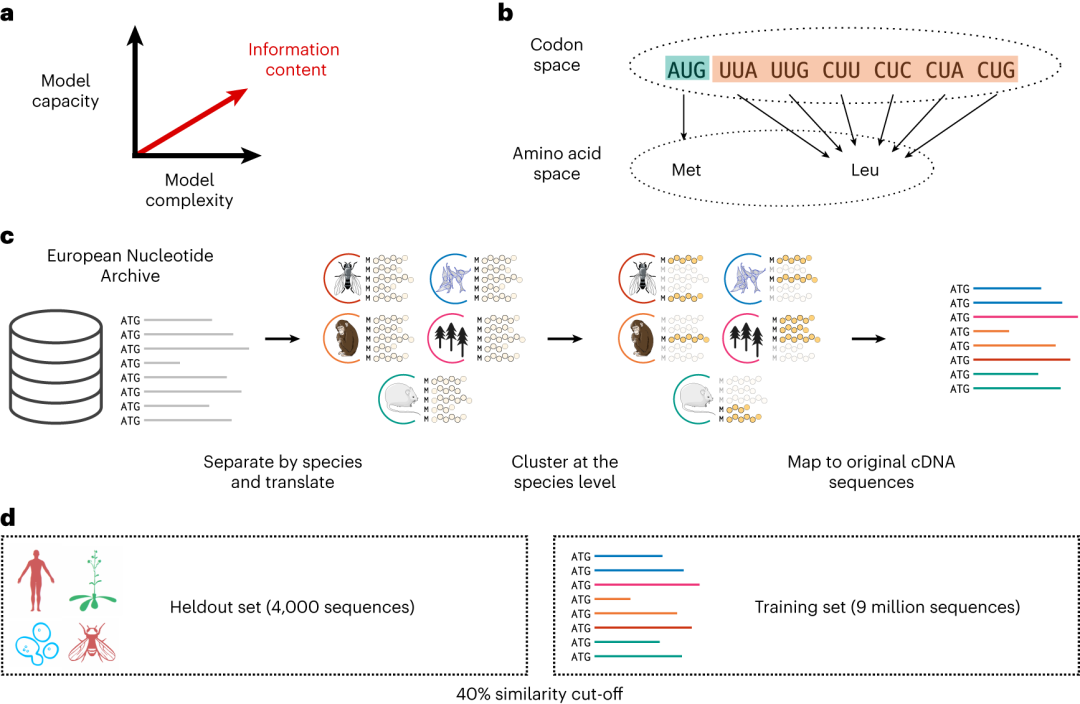
图示:将蛋白质语言模型扩展到密码子语言。(来源:论文)
密码子的使用也与蛋白质折叠有关,有充分的证据表明密码子序列的变化会影响折叠动力学、折叠途径,甚至正确折叠的蛋白质的量。这一证据表明,同义密码子的使用包含有价值的生物信息,机器学习模型可以利用这些信息来提高预测任务中的信噪比。
用密码子序列,而不是氨基酸序列
在最新的研究中,牛津大学的研究团队证明在密码子序列上预训练蛋白质语言模型 CaLM(codon adaptation language model,由 8600 万参数进行训练),可以产生能够捕获关键生化特征的信息丰富的蛋白质表征。测试表明,根据密码子而不是氨基酸序列训练的蛋白质表征,在各种下游任务中表现出显著的优势。
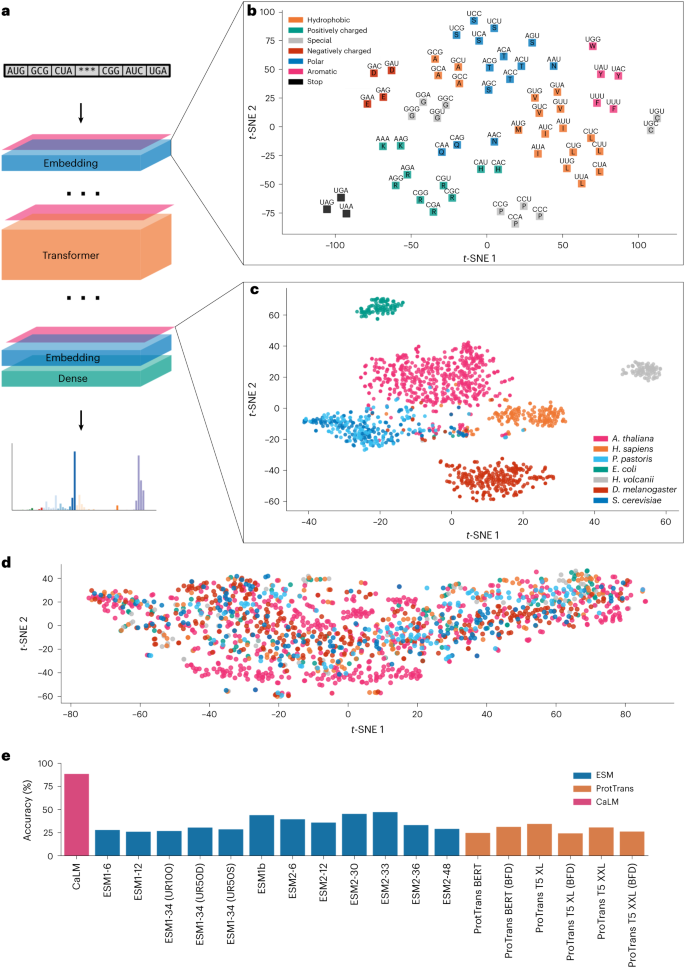
图示:CaLM 概述。(来源:论文)
该团队的 8600 万参数语言模型的性能,优于其他具有类似容量的模型,在许多情况下,甚至优于参数超过 50 倍的模型。这种性能是由于密码子语言模型能够捕获跨 DNA 序列的密码子使用模式的能力,并且当密码子使用信息被损坏时,这种优势就会消失。
cDNA 训练模型的额外训练成本可以忽略不计,并且似乎可以提高所考虑的所有序列级任务的性能。由于高通量蛋白质测序几乎完全是通过 DNA 序列的翻译来完成的,因此原始编码序列是公开可用的并且可以用于训练。研究人员建议使用 cDNA 而不是简单的氨基酸序列来训练蛋白质语言模型,这为改进计算蛋白质工程提供了一条明确的途径。
密码子语言模型还可以为无需比对的蛋白质结构预测,提供有价值的进化信号,特别是在依赖语言模型来预测蛋白质各部分之间关系的 ESMfold 和 OmegaFold 等方法中。
基于 cDNA 的模型可以恢复更广泛的进化关系,例如同义突变,这在核苷酸水平上很明显,但在氨基酸水平上并不明显。已知同义密码子的使用与结构特征相关,并且密码子使用和蛋白质折叠之间的联系可能为已知无法捕获折叠物理原理的方法提供有价值的信号。
研究人员建议,将密码子语言模型纳入免比对蛋白质结构预测的流程中,很可能为加速高精度蛋白质结构预测提供一条成本可以忽略不计的途径。
提高蛋白质表达质量的两个方向
该团队还提出了进一步提高蛋白质表达质量的两个主要方向。
一是规模扩大。当前的研究使用了一个只有 8600 万个参数的简单模型,这个大小与最新出版物中的标准模型大小相比显得相形见绌。
使用的数据集也相对较小:与 ESM 系列模型中使用的 1.25 亿个序列或某些 ProtTrans 模型中使用的近 5 亿个序列相比,仅 900 万个序列。通过在包含数亿 DNA 序列的数据集上训练数十亿参数模型,存在一条明确的途径来提高表征质量。
另一个潜在的改进方向是开发结合氨基酸和编码序列的多模式模型。该研究的消融实验表明,在缺乏密码子使用信息的情况下,模型性能大幅下降,以至于低于数据集中的每个氨基酸模型。然而,由于模型间接访问氨基酸序列,因此原则上它应该能够访问与仅氨基酸模型相同的信息。
这种差异可能是由于训练期间缺乏氨基酸水平信号造成的,因此结合氨基酸和密码子序列的训练模型可以提高整体模型性能。
更丰富的输入带来新视角
在生物学中,人们非常关注数据集偏差的影响,但相比之下,人们很少甚至没有关注蛋白质工程中更丰富的输入的重要性。随着计算能力和模型架构的进步,利用更丰富的生物数据为提高生物学中机器学习的能力提供了明确的方向。
基于 cDNA 训练的大型语言模型的开发,将使研究「不直接由氨基酸序列确定的蛋白质特性」成为可能。例如,密码子的使用与蛋白质折叠的相关性,实验证据表明密码子序列的变化确实会影响折叠动力学、折叠途径,甚至正确折叠蛋白质的数量。
仔细选择密码子序列是蛋白质科学的一个关键目标,其中表达的 cDNA 的特定序列会对产量产生巨大影响。该团队提出的基于密码子的语言模型,代表了使用机器学习方法来研究蛋白质的这些特性和其他特性的第一步,而这些特性迄今为止还没有被氨基酸语言模型解决。
相关报道:https://www.nature.com/articles/s42256-024-00791-0